超详细盘点!详解体内 CAR-T 疗法的最新载体技术
编译丨Aspen
来源丨金斯瑞
嵌合抗原受体 T 细胞 (CAR-T) 治疗是一种特殊的癌症免疫疗法:它拥有前所未有的疗效,但研发过程复杂且成本高企。到目前为止,该疗法还不是一个能让所有患者都受益的治疗选择。这促使全世界科研人员共同努力来改进 CAR-T 疗法。
在所有已提出的改进方案中,直接在患者体内生成 CAR-T 细胞可以说是最具挑战性、也最具临床价值的策略。这可以使 CAR 疗法从一种基于细胞的自体药物产品转化成为一种具有普适性的现货疗法。

近日,MolecularTherapy 期刊发表了一篇综述论文,介绍了 目前最前沿的体内 CAR 治疗方案,着重盘点了这些疗法中所使用的载体技术。这些载体包括慢病毒载体 (LV) 、腺病毒相关载体 (AAV) 、合成聚合物纳米载体 (NC) 和脂质纳米颗粒 (LNP) 。相关技术的概念证明都已经在小鼠体内完成。
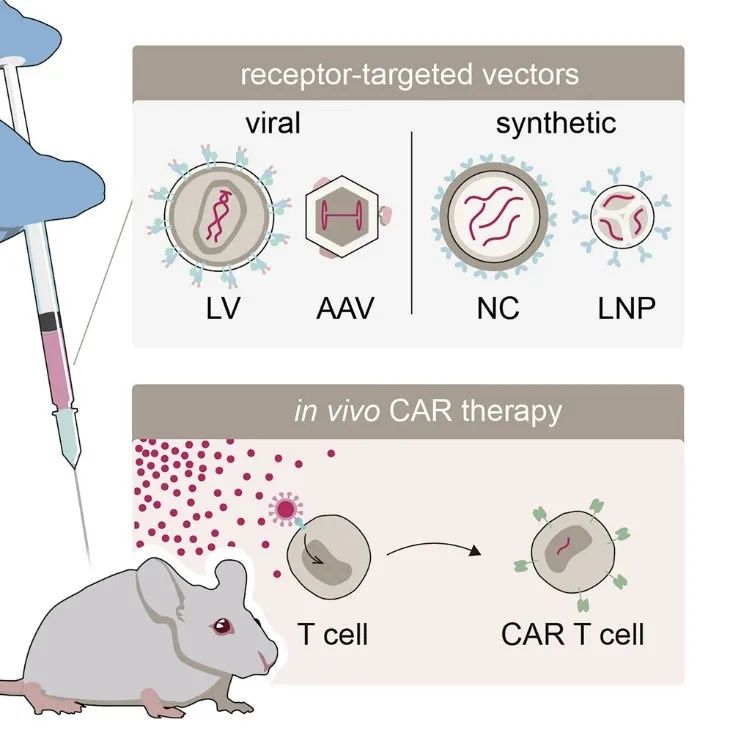
载体颗粒的受体靶向是至关重要的,因为它可以防止 CAR 基因传递到非靶向细胞,从而降低脱靶毒性。因此,这篇文章还 讨论了不同载体平台的特性,例如它们的免疫原性、效力和 CAR 递送模式 (永久的还是瞬时的) 等等。
最后,Alexander Michels 等人概述了将体内 CAR 治疗从概念验证推进到可用于临床试验的强大且可扩展的技术所需做的工作。
特别说明:金斯瑞团队对这篇综述进行编译,因原文篇幅过长。本篇以体内 CAR-T 疗法载体技术相关内容为主,其余体内 CAR-T 技术现状及未来展望相关内容将在下一期文章中发布。
基因疗法
基因疗法发展至今已有30余年历史,伴随着本世纪初的成功案例之后,许多关于基因疗法副作用和死亡案例的报道接踵而至。随后,学者们通过载体设计方面的技术创新克服了这些挑战。随着学术实验室,生物技术公司和大型制药公司的纷纷入局,基因治疗领域进入了蓬勃发展阶段。
这一发展的主要驱动力是从单基因疾病到复杂的获得性疾病 (如癌症) 的适应症的扩展,而编码人造蛋白质的基因工程也促进了这一领域的发展。其中最突出的一个例子就是 CAR,目前已有数款 CAR 产品获批上市,彻底革新了癌症免疫治疗领域。
尽管如此,基因治疗的主要技术挑战仍然存在:除了在单基因疾病治疗中实施微创基因编辑和提高治疗安全性以外,治疗基因在体内的细胞选择性递送是目前研究的重点之一。直接在患者体内将治疗相关细胞转化为能产生修复或治疗性蛋白质的细胞,将使我们绕开复杂的基因修饰细胞的体外制造,并使以前不可行的治疗方法成为可能。

体内和体外疗法的比较
目前,体内基因传递已应用于临床治疗。基于AAV 或溶瘤病毒,这些疗法依赖于其递送物质的局部应用性或非毒性来抵消载体广泛的细胞趋向性。然而,对于许多适应症,治疗药物的脱靶递送现象存在着安全隐患。CAR-T 细胞治疗也是如此。
体内 CAR-T 疗法
CAR-T 细胞疗法是过继性T细胞疗法的一种,在这种治疗中,癌症患者接受经过基因改造和体外扩增的肿瘤特异性的T细胞。CAR 是由一个胞外抗原结合域,通过铰链区和跨膜域连接到一个或多个细胞内信号域而形成的。在与目标细胞结合后,CAR 会诱导细胞内信号传导,导致 CAR-T 细胞增殖,同时特异性杀伤目标细胞。因此,CAR-T 细胞可以被认为是一种活体药物,当它们遇到目标癌细胞时,会在患者体内放大。
近年来,这种独特的治疗理念推动了全球研究,目前有六种 CAR-T 产品获美国 FDA 批准,有四种 CAR-T 产品已在欧洲获得上市许可。最早获批上市的是 Yescarta 和 Kymriah,这两款药物都通过 B 细胞标记 CD19 靶向淋巴瘤细胞。除了淋巴瘤细胞外,健康的B淋巴细胞也会被这些产品消除,因此B细胞耗竭是受治患者最突出的副作用。
最近,另外两款产品 Tecartus 和 Abecma 也已获得监管部门的批准,后者通过 B 细胞成熟抗原 (BCMA) 将 CAR 治疗的适应症扩展到多发性骨髓瘤。首个非靶向 CD19 的 CAR 细胞产品的批准标志着癌症免疫治疗这一新领域的又一次重大突破。事实上,全世界正在进行数百项 CAR-T 细胞试验,其中许多试验旨在研发新靶点和治疗其他恶性肿瘤。
作为一种创新疗法,CAR-T 细胞在病人身上的使用已经成为了一场关于医疗系统成本问题的公开辩论。CAR-T 细胞十分昂贵,因为它们是个性化的细胞治疗产品,需要依赖于体外基因转移方案的漫长的制造过程。在分离患者的淋巴细胞后,细胞被激活并随后被转导,其载体通常为慢病毒或γ逆转录病毒。然后,经过修饰的淋巴细胞被扩增,最后重新注入患者体内。这种复杂的制造过程是昂贵的。
此外,T 细胞必须严格从患者的血液中分离出来,因为在转导过程中残留的肿瘤细胞可能会导致 CD19-CAR 在制造过程中意外转移到白血病细胞中。这会导致 CAR-T 细胞抗性克隆的产生,其中 CD19 被 CAR 的单链可变抗体片段 (scFv) 掩盖,从而无法被正确识别。患者会因此复发癌症或死亡。虽然到目前为止只报道了一例这样的病例,但它强调了与 CAR-T 脱靶转移相关的风险,突出了 CAR-T 细胞制造过程的复杂性。这阻碍了它们在标准医疗保健中的广泛应用。
因此,目前在临床前和临床研究中正在寻求各种途径来改进 CAR 技术。从自动化系统到异体 CAR-T 细胞,学者们提出了一系列旨在优化生产过程的策略。虽然自动化方案结合了在病床边生成 CAR-T 细胞的可能性,这将大大促进生产与药物分发,但这不会改变产品自体、高度个性化的本质。
在异体方法中,从健康供体制备的CAR-T细胞经过基因处理,可以降低其在受体中的异体反应性。这也就是我们常说的 “现货” (off-the-shelf) CAR-T。但是最终的 “现货” 产品很可能不会完全通用,因为人类白细胞抗原屏障需要适应患者亚群。当 CAR-T 细胞直接在体内产生时,可以规避移植物与宿主间的反应以及制造相关的复杂问题。在体内 CAR-T 疗法中,一个单一的,普遍适用的药物产品,编码 CAR 的系统递送载体将被直接用于转导患者的T细胞。因此,这个过程产生的 CAR-T 细胞将是真正的自体细胞。尽管从理论上来说,这是满足 CAR 治疗日益增长的需求的理想方法,但体内方法还没有进入临床,主要是因为缺乏合适的载体平台。

自体疗法和异体现货疗法
但是,现在情况已经有所改变。在过去的5年里,有几个研究小组已经报道了在小鼠模型中成功完成体内 CAR-T 细胞生成的案例。伴随着载体设计的发展,特别是在载体靶向方面的进步,学者们充分说明了载体工程在基因治疗中的重要性。这些载体研究应被看作是癌症免疫治疗领域的重要突破。
受体靶向基因递送载体
尽管已有数款药物获批,临床研究也取得了突破,但是基因疗法远未到达广泛运用的程度。已获批的产品可应对严重疾病,其中与治疗相关的风险是可以接受的。但随着基因疗法开始被考虑用于更多的适应症,改进载体技术的需求也就越来越突出。
这与全身给药尤其相关,全身给药导致潜在的剂量限制性毒性,包括但不限于肝毒性,这点已在某些脂质纳米颗粒 (LNP) 和 AAV 研究中阐明。在一项研究 AAV8 治疗 X 连锁肌管肌病的试验中,所有三名接受较高剂量的患者都经历了严重的肝毒性,其中两人最终死亡。
两名接受 Zolgensma 治疗的已故儿童的人类生物分布数据表明,将 CAR 靶向肝脏以外的器官时需要高载体剂量,因为只有一小部分被注射的颗粒会到达与治疗相关的细胞 (Zolgensma是一种基于AAV9的基因疗法,用于治疗脊髓性肌萎缩症) 。
在全身给药时优先(甚至完全)转导治疗相关细胞类型的载体有望减少所需的剂量和毒性。对于体内 CAR 治疗而言,使用这种T细胞靶向载体有望防止 CAR 递送到非 T 细胞,避免对患者造成致命后果。对于任何新型的T细胞靶向载体,T细胞选择性的程度必须通过实验来确定。受体靶向是一种合理的载体设计方法,它可以使载体对特定的细胞表面受体具有选择性,例如 CD3、CD4、CD5 或 CD8 等免疫细胞标记物。重要的是,选择性是在细胞进入的水平上实现的。因此,这些载体优先转导在其表面显示目标受体的细胞。对感兴趣的器官的一些选择性可以通过 AAV 衣壳中纳米颗粒或残基的化学组成来实现。另外,在受体靶向过程中,将高亲和力的结合物,如 scFv或设计的锚蛋白重复蛋白 (DARPins) 纳入颗粒结构,可以产生接近绝对的细胞类型选择性 (95%) ,这一点在慢病毒载体 (LV) 实验中尤其明显。

不同载体平台技术路径比较
慢病毒载体
慢病毒载体通常为水疱性口炎病毒 (VSV) 糖蛋白 G 的假性分型,颗粒直径为120-150 nm。VSV 糖蛋白 G 通过在多种细胞类型上表达的低密度脂蛋白受体 (LDLR) 及其家族成员介导细胞进入。因此,VSV-G 型慢病毒表现出广泛的趋向性,在不同的人类细胞类型上实现高效率转导,其中包括活化的 T 淋巴细胞。CAR 治疗主要依赖于慢病毒或γ逆转录病毒载体的体外转导。在2018年,美国54%的临床研究使用慢病毒来生成 CAR-T 细胞。除此之外,超过100个已注册的慢病毒临床试验正在开展,其中大多使用慢病毒来治疗免疫和血液系统疾病或癌症。
用于受体靶向的慢病毒合并包膜蛋白工程分为两步:1)破坏自然受体的使用;2)添加用于目标识别的结合子。有学者正在尝试改变 VSV G 蛋白受体的使用,但这一研究颇具挑战性。因为 VSV G 蛋白受体介导受体结合和膜融合。尽管最近对 LDLR 接触残留物的鉴定已经促进了靶向受体 VSV-LV 在细胞相互作用研究中的使用,但是依靠α病毒和副粘病毒的糖蛋白来做慢病毒靶向的方法是更先进的,因为它们有单独的包膜蛋白来做结合和融合。该方法最初是针对麻疹病毒 (MV) 糖蛋白开发的,而后扩展到尼帕病毒 (NiV) 的包膜蛋白。当膜近端受体域被靶向时,NiV-LV 表现出比 MV-LV 更高的载体滴度,并且对人血清抗体具有耐药性。
本文作者所在的实验小组在慢病毒靶向方面做了很多开创性的工作,他们发现了十多种对不同的细胞表面蛋白具有特异性的慢病毒。除了肿瘤细胞、神经亚型和内皮细胞的表面标记物外,这些研究还包括与体内 CAR 基因传递高度相关的免疫和造血细胞标记物。第一个被工程化副粘病毒蛋白成功靶向的T淋巴细胞标志物是 CD8。有趣的是,这项研究表明,慢病毒的受体靶向不仅能介导靶细胞的高选择性,还能将激活刺激传递给靶细胞。使用来自已激活 CD3 抗体的单链抗体会激活静息 T 淋巴细胞并直接在未加工的血液中实现基因转移。
另一个重要的方面是靶标受体下调 (以人类CD4靶标为例) 。在加入载体颗粒后的几天内会检测到靶标受体表达下调的现象,但这种下调往往是短暂的。随着靶标受体表达的完全重建,靶标表达会恢复正常。然而这种瞬时性受体下调是否对体内 CAR 治疗具有功能性后果仍有待研究。
通过甲病毒来源的进入机制,特别是Sindbis病毒 (SINV) 的包膜蛋白靶向的慢病毒,在工程方法上与基于副粘病毒糖蛋白的细胞进入模式不同。在一种方法中,SINV E1 糖蛋白与选择性决定分子 (如完整的CD3特异性抗体) 一起显示在慢病毒表面上。在另一种更常用的策略中,SINV E2 受体结合蛋白的自然趋向性通过突变被消除,载体颗粒通过与 E2 和细胞靶受体同时相互作用的分子适配子而被重定向到选择的受体上。在其中一种策略中,研究人员使用含有与来自蛋白质 A 的 Fc 结合 ZZ 结构域的 E2,然后选择抗体 (例如针对CD4的抗体) 与之结合。其他基于适配子的策略也会使用 SpyTag-SpyCatcher 或二硫键形成的蛋白质-肽对,生物素化抗体,或串联 Fab 作为适配子。这些策略加强了适配子的稳定性,尤其适合体内应用。
副粘液和α病毒假性类型之间的一个关键区别是它们的 pH 依赖性。SINV E1 依赖于内含体中的低 pH 值来介导膜融合和细胞进入。因此,以 SINV 为基础的靶向 CD4 的慢病毒,其转导会被 dynamin 突变和酸化抑制剂所抑制。与此形成鲜明对比的是,基于副粘液的 Her2/neu-或 EPCAM 靶向 MV 和 NiV-LV 会在不受 pH 影响的情况下与细胞膜融合。当内体酸化被阻断时,转导的强烈增加证明了这一点。因此,它们也可以通过不经常内吞的受体来转导细胞。所以,副粘菌假型慢病毒的靶细胞选择性可能接近100%。尽管有许多研究都在报告蛋白水平上的高选择性,但最近的一项研究应用单细胞转录组学研究了人类外周血单个核细胞中靶向人类 CD8 的慢病毒的靶细胞选择性。这项详尽的研究揭示其在靶效率超过99%。少数被归类为脱靶的细胞在载体颗粒进入时,其靶受体是否呈阴性仍有待研究。
腺相关病毒载体
腺相关病毒载体 (AAV) 在很多方面与LV截然不同。AAV 由包含单链 DNA 基因组的非包膜蛋白衣壳组成,因此它的遗传修饰通常是短暂的,特别是在增殖细胞中 (如活化的淋巴细胞) 。在与它们的靶受体相互作用后,网格蛋白介导内吞作用和胞内运输 (这些过程的细节由衣壳的血清型决定) ,AAV 进入细胞核并释放单链转基因,在第二链合成后,该转基因可被用于转录。
由于缺乏膜状包膜,AAV 的结合子必须被整合到坚硬的衣壳中,以便完成受体靶向。从VP1 到 VP3,不同的衣壳蛋白共享 VP3 的公共区域,但其灵活的 N 端尾部却不尽相同。由质粒产生的 AAV 衣壳按照随机抽取的原则随机组装,在给定的制备条件下,几乎每个衣壳都具有 VPs 的独特化学空间配置。同样,AAV 的结合和细胞摄取也是一个复杂且具有血清型依赖性的过程。AAV 通常使用聚糖作为附着因子,如 AAV2 中使用的硫酸乙酰肝素蛋白聚糖 (HSPG) 。此外,数十种细胞蛋白与 AAV 摄取有关,其中包括跨膜 AAVR 受体,这对大多数血清型至关重要。
AAV 的衣壳制作通常是基于对载体颗粒库的选择,这些载体颗粒库通过重组血清型或插入组合多样化的多肽来生成。虽然学者们基于这个概念生成了许多新型衣壳,这些衣壳具有优先分布到靶组织的特性,但是利用这种方法所获得的载体颗粒通常缺乏完全的细胞型选择性。因此,想要让 AAVs 获得与前文慢病毒相似的对 T 淋巴细胞的特异性,这种方法是行不通的。在慢病毒中获得原理证明的方法是在 VP2 的 N 端展示针对肿瘤标记物 Her2neu 的 DARPin。为了消除 AAV2 衣壳与 HSPG 的结合,研究人员突变了两个相关的精氨酸残基。值得注意的是,靶向大大减少了静脉内给药载体在肝脏的积累。同样,AAVs 也被生成用于特异性转导 GluA4-、CD105-、EpCAM- 和 CD4 阳性细胞。这些细胞都被成功地运用于小鼠模型或人类血液实验中。
在 N 端靶向方法的基础之上,通过蛋白质剪接可以将 DARPin 和 scFvs 作为蛋白质共价物偶联到通用 AAV 的衣壳上。也有学者将光敏色素相互作用因子6与 VP2 耦合形成光诱导转导系统。有趣的是,在 65°C 下孵育载体储液可提高转导效率。这可能表明并非所有的N端融合结合子都位于衣壳之外,而衣壳中由热引起的构象变化将这些以前无法接触到的结合子暴露在颗粒表面。有学者通过筛选用于衣壳组装和基因传递的 mCherry 插入文库,在 VP3 共有区 GH2-GH3 环上确定了一个表面暴露的插入位点。Eichhoff 等人将该插入位点运用于纳米抗体,成功地将载体靶向 CD38,ARTC2.2 和 P2X7。另外,将 DARPin 插入 VP1 GH2-GH3 环可以产生靶向小鼠 CD8 的 AAV。在小鼠原代脾细胞上,产生的 mCD8-AAV 的转导效率比亲本 AAV2 高26倍,其中99% 的绿色荧光蛋白阳性细胞都为 CD8+。出人意料的高基因传递活性使得这种被称为 DART-AAV 的新型载体与体内 CAR-T 细胞生成高度相关。
非病毒载体
随着基于 mRNA 的新冠疫苗在世界范围内的运用,非病毒载体 (尤其是LNP,数十项相关临床实验正在进行) 近期获得了全世界的关注,将它们用于基因治疗的研究也正在蓬勃发展。非病毒载体颗粒不像病毒载体颗粒那样有序。与复杂的病毒机制不同,非病毒载体的细胞进入、运输和转基因递送等过程都是由粒子和其有效载荷的理化性质介导的。
除了 LNP 之外,纳米载体 (NC) 也已被用于体内 CAR 递送。NC 是带负电荷的核酸和带正电荷的聚 (β-氨基酯)(PBAE) 聚合物的复合物。它们被细胞内吞吸收。被细胞摄取后,NC 的核酸会从内含体中逸出,从而进入细胞。已有学者报道含有质粒或者体内转录 mRNA 的 NC。质粒 DNA 可通过与 PBAE 载体材料共价连接的核定位-微管相关信号肽 (NLS-MTAS) 定向到细胞核。当转座酶 (如PiggyBac) 的表达质粒与转基因质粒共传递时,后者可以整合到宿主细胞的染色质中。
在 LNP 中,mRNA 通过静电相互作用被封装在脂质颗粒的核心部位。在完成宿主细胞摄取和内体逃逸过程后,mRNA 可用于翻译,直到它被降解。为了解决 RNA 生物稳定性和免疫原性问题,研究人员可能会进行一些修饰:这些修饰包括添加 5' 帽类似物和 3' poly(A) 尾,在 5' 和 3' 非翻译区 (UTR) 中引入稳定序列,转录的密码子优化和使用免疫原性较低的修饰核苷酸,例如 N1-甲基假单核苷酸。
阐明病毒和非病毒载体之间的根本区别,对非病毒载体进行用于受体靶向的蛋白结合物修饰 (如抗体或fab) ,这些都是有机化学的研究课题。当然,偶联的细节取决于所讨论的纳米颗粒的组成,已有学者提供了这方面可用策略的详细综述。最近在靶向 mRNA-LNP 方面取得的成功依赖于 SATA-马来酰亚胺的化学性质。在这里,学者将含有用马来酰亚胺功能化的聚乙二醇脂质衍生物的胶束 (DSPE-PEG-mal) 与已经形成的 mRNA-LNP 混合以将 DSPE-PEG-mal 插入 LNPs 中。然后,通过使用 N-琥珀酰亚胺基S-乙酰硫代乙酸酯 (SATA) 在伯胺上引入受保护的巯基,使抗体功能化。脱保护后,抗体通过在巯基和马来酰亚胺基团之间形成硫醚键与 LNPs 完成结合。使用这种方法, mRNA 已被特异性递送到 PECAM-1,CD4 和 CD5 阳性细胞中。
对于 NC,静电相互作用对于靶向配体的偶联至关重要。为此,聚谷氨酸 (PGA) 在与抗体或 Fab 偶联之前先用乙基-N-(3-二甲氨基丙基) 碳二亚胺活化。带负电的 PGA 抗体偶联物通过静电相互作用与 DNA 和 PBAE 的带正电复合物结合。应当注意的是,当使用如上所述的化学共轭方法时,有些粘合子在颗粒表面上的方向有可能是错误的。
参考论文:
[1] Michels, Alexander, Naphang Ho, and Christian J. Buchholz. "Precision Medicine: In Vivo CAR Therapy as a Showcase for Receptor-Targeted Vector Platforms." Molecular Therapy (2022).
相关阅读
-

在 LNP 中,mRNA 通过静电相互作用被封装在脂质颗粒的核心部位。在完成宿主细胞摄取和内体逃逸过程后,mRNA 可用于翻译,直到它被降解。...
-

极目新闻记者 康旭阳通讯员 刘汉生提起古生物化石,你是否会觉得,这些生活在千万年前、数亿年前的古生物,距离我们很遥远?虽然古生物生活的年代离我们非常远,但是...
-

我不想再过“手心向上”的卑微生活,适合全职妈妈的副业,从0到1
在我家萱宝8个月的时候,我去衣柜放衣服,把孩子顺手放在床边上,只听“咚”一声,孩子滚落下来,孩子哭的撕心裂肺,我眼看着头上鼓出个大鼓包,心里真是...
-

【干货】疫苗生产概念股查询_附:股票名单(2024/6/13)
【干货】疫苗生产概念股查询_附:股票名单(2024/6/13),据南方财富网概念查询工具数据显示, 疫苗生产概念股有: 1、万泰生物: 回顾近7个交易日,万泰生物有3天上涨。...
-

十大公认最好看的电视剧,20年的经典难超越(刷过N遍的电视剧)
每一年都会上新很多的电视剧,这些电视剧在题材、演员、质量上都是有很大的区别,好看的电视剧就算是过了很长时间也是会拿出来回味一下的,流量的电视剧即使是自己很喜欢的演...
-
极目新闻记者 康旭阳通讯员 刘汉生提起古生物化石,你是否会觉得,这些生活在千万年前、数亿年前的古生物,距离我们很遥远?虽然古生物生活的年代离我们非常远,但是...
-

营养缺乏与营养过剩,都会削弱免疫功能。ω-3脂肪酸深海鱼类如鲑鱼、沙丁鱼、鲔鱼、鲭鱼,坚果类如亚麻籽、核桃等➤ω-3和ω-6多不饱和脂肪酸,特别...
-

你认为世界上什么最坚硬?盔甲,钢铁,金银?普通人认为这些已经足够抵挡刀剑了,但是如果是遇上猛兽呢?遇到狮子老虎这样牙尖嘴利的猛兽,这些金银钢铁制品恐怕会变得不堪一击,可能很多......
-

藏不住了!太子山里的临夏宝藏美景,太子山,临夏,雪峰,莲花山,康乐县,松鸣岩...
-
JAMA杂志上一项研究初步发现,代谢功能障碍相关脂肪变性肝病(MASLD)患者,每天81 mg阿司匹林能显著减少肝脏脂肪量。这是一项为期6个月的...
-

在过去的二三十年里,全球肥胖及其相关代谢紊乱的发生率呈指数级增长,大大增加了2型糖尿病、非酒精性脂肪肝、动脉粥样硬化、心血管问题和一些类型的癌症...
发表评论
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任如发现本站有涉嫌抄袭侵权/违法违规的内容,请发送邮件举报,一经查实,本站将立刻删除。
